Στον καρκίνο του παχέος εντέρου το 95% αφορά σποραδικές περιπτώσεις ενώ στο 5% υπάρχει κάποιου είδους κληρονομικότητα που μπορεί να μεταβιβάζεται με επικρατή ή υπολειπόμενο χαρακτήρα. Το πιο συχνά παρατηρούμενο σύνδρομο είναι το Lynch που χαρακτηρίζεται από καρκίνους του παχέος εντέρου, του ενδομητρίου και διάφορους άλλους όγκους (1). Το σύνδρομο αυτό προκαλείται από μεταλλαγές στα γονίδια επιδιόρθωσης του DNA (MLH1, MSH2, MSH6 και PMS2). Η οικογενής αδενωματώδης πολυποδίαση παρατηρείται στο 1% των περιπτώσεων καρκίνου του παχέος εντέρου (2) και χαρακτηρίζεται από την ανάπτυξη εκατοντάδων έως χιλιάδων αδενωματωδών πολυπόδων στο παχύ έντερο ενώ συχνά παρατηρούνται και εξωεντερικές εκδηλώσεις της νόσου. Η πιθανότητα κακοήθους εξαλλαγής κάποιου εκ των πολυάριθμων πολυπόδων και ανάπτυξης καρκίνου σε νεαρή ηλικία αγγίζει το 100% (3).
Στο 8% των οικογενειών περιγράφεται μια ήπια μορφή του συνδρόμου με ανάπτυξη μικρότερου αριθμού πολυπόδων στο παχύ έντερο και σε μεγαλύτερη ηλικία (attenuated FAP) (4). Το σύνδρομο της οικογενούς αδενωματώδους πολυποδίασης μεταβιβάζεται κατά τον αυτοσωμικό επικρατή χαρακτήρα και προκαλείται από μεταλλαγές του γονιδίου APC (Adenomatous Polyposis Coli), το οποίο παίζει σημαντικό ρόλο στην κυτταρική αύξηση. Σχετικά πρόσφατα ανακαλύφθηκε ένα επιπλέον γονίδιο του οποίου οι μεταλλαγές προκαλούν πολυποδίαση, το MUTYH, και η πολυποδίαση που σχετίζεται με το συγκεκριμένο γονίδιο μεταβιβάζεται με τον αυτοσωμικό υπολειπόμενο χαρακτήρα (5).
Οικογενής αδενωματώδης πολυποδίαση (Familial adenomatous polyposis – FAP)
Η οικογενής αδενωματώδης πολυποδίαση έχει επίπτωση 1 ανά 10000 γεννήσεις και προκαλείται από μεταλλαγές του γονιδίου APC. Το 15-20% αυτών αφορούν πρωτοεμφανιζόμενες περιπτώσεις καθώς δεν εντοπίζονται σε προηγούμενες γενεές (6). Η κλινική εκδήλωση του συνδρόμου περιλαμβάνει την ανάπτυξη εκατοντάδων πολυπόδων στο παχύ έντερο κατά τη διάρκεια της παιδικής και εφηβικής ηλικίας. Εάν δεν αντιμετωπισθούν χειρουργικά θα αναπτυχθεί καρκίνος του παχέος εντέρου μέχρι την ηλικία των 40-50 ετών.
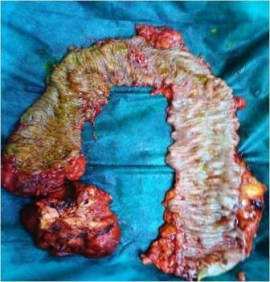
Στο 8% των ασθενών εμφανίζεται μια ηπιότερη μορφή του συνδρόμου με μικρότερο αριθμό πολυπόδων στο έντερο και καθυστερημένη ηλικία εμφάνισης αυτών. Στο 5% περίπου των ασθενών αναπτύσσονται και πολύποδες στο ανώτερο γαστρεντερικό σύστημα και κυρίως στο δωδεκαδάκτυλο, οι οποίοι εάν δεν αντιμετωπισθούν μπορούν να οδηγήσουν σε καρκίνο. Στο 10-15% των περιπτώσεων αναπτύσσονται δεσμοειδείς όγκοι που αν και είναι καλοήθεις δύναται να συμπεριφέρονται επιθετικά λόγω αύξησης του όγκου τους και πίεσης παρακείμενων ζωτικής σημασίας ιστών. Οι εξωεντερικές εκδηλώσεις του συνδρόμου της οικογενούς αδενωματώδους πολυποδίασης συνοψίζονται στον Πίνακα 1.

Πίνακας 1.
Εξωεντερικές εκδηλώσεις οικογενούς αδενωματώδους πολυποδίασης
Η βασική διάγνωση του συνδρόμου γίνεται κλινικά με την ανεύρεση περισσοτέρων από 100 πολυπόδων στο παχύ έντερο των ασθενών. Όσον αφορά την κλινική διάγνωση της ηπιότερης μορφής του συνδρόμου, εδώ υπάρχουν μεγαλύτερες δυσκολίες. Πρόσφατα διαγνωστικά κριτήρια περιλαμβάνουν: α) τουλάχιστον 2 ασθενείς με 10-99 πολύποδες σε ηλικία άνω των 30 ετών, β) 1 ασθενής με 10-99 πολύποδες σε ηλικία άνω των 30 ετών και 1 πρώτου βαθμού συγγενής με καρκίνο του παχέος εντέρου και λίγους πολύποδες. Άλλοι ερευνητές έχουν προτείνει (10): α) κληρονομικότητα με επικρατή χαρακτήρα, β) παρουσία 3-99 πολυπόδων σε ηλικία άνω των 20 ετών.
Στο 70% των ασθενών με τυπική οικογενή αδενωματώδη πολυποδίαση ανιχνεύονται μεταλλαγές του γονιδίου APC, ενώ στην ηπιότερη μορφή του συνδρόμου το ποσοστό φθάνει μόλις το 25% (4). Σε όλους τους ασθενείς πρέπει να δίνεται γενετική καθοδήγηση καθώς όταν ανιχνευθεί η μεταλλαγή στο γονίδιο APC πρέπει να ακολουθεί προληπτικός έλεγχος στους πρώτου βαθμού συγγενείς. Τα μέλη της οικογένειας που εντοπίζονται να φέρουν τη μεταλλαγή πρέπει να εντάσσονται σε πρόγραμμα παρακολούθησης από την εφηβική ηλικία.
Στον Πίνακα 2 συνοψίζονται οι κατευθυντήριες οδηγίες αντιμετώπισης τόσο του τυπικού συνδρόμου όσο και της ήπιας μορφής αυτού.

Πίνακας 2:
Πρωτόκολλο παρακολούθησης του εντέρου σε ασθενείς με οικογενή πολυποδίαση
Η εκτομή του παχέος εντέρου με πολυποδίαση πριν οι πολύποδες εξαλλαχθούν είναι ιδιαίτερα σημαντική καθώς μειώνονται η νοσηρότητα και θνητότητα που σχετίζονται με την ανάπτυξη καρκίνου. Όσον αφορά την επέμβαση εκλογής για τους ασθενείς με πολυποδίαση πρέπει να σημειωθεί ότι οι δύο κύριες επιλογές είναι από τη μια μεριά η αφαίρεση του παχέος εντέρου και ειλεο-ορθική αναστόμωση και από την άλλη η πρωκτοκολεκτομή με ειλεο-πρωκτική αναστόμωση και δημιουργία ειλεϊκής ληκύθου. Η πρώτη επέμβαση είναι σαφώς πιο εύκολη από τη δεύτερη, η οποία είναι τεχνικά πολύ πιο απαιτητική. Οι επιπλοκές σπανίζουν και η κινητικότητα του εντέρου μετεγχειρητικά είναι πολύ καλή. Η εκτεταμένη χειρουργική προσπέλαση της δεύτερης επιλογής έχει τον κίνδυνο της αιμορραγίας, της κάκωσης των πυελικών νεύρων και της υπογονιμότητας (7).
Η πρωκτοκολεκτομή πρέπει να επιλέγεται όταν υπάρχει μεγάλος αριθμός (>15-20) αδενωμάτων στο ορθό. Αντίθετα, όταν οι πολύποδες στο ορθό είναι λίγοι ή λείπουν εντελώς τότε και οι δύο επιλογές είναι δυνατές. Το είδος της μεταλλαγής στο γονίδιο APC παίζει καθοριστικό ρόλο στον αριθμό των πολυπόδων του εντέρου. Σε μελέτη ανασκόπησης πάνω στο θέμα αυτό βρέθηκε ότι μεταλλαγές στα κωδικόνια 1250-1464 του APC και ειδικότερα στο κωδικόνιο 1309 σχετίζονται με σοβαρή νόσο, ενώ αντίθετα μεταλλαγές στα ακραία κωδικόνια του γονιδίου και σε θέσης εναλλακτικού ματίσματος του εξωνίου 9 οδηγούν σε ήπιο φαινότυπο (8). Τα παραπάνω οδήγησαν στην πρόταση να γίνεται επιλογή της χειρουργικής επέμβασης με βάση το γενετικό έλεγχο αυτών των ατόμων. Έτσι, ασθενείς με αναμενόμενη σοβαρή νόσο πρέπει να υποβάλλονται σε πρωκτοκολεκτομή ώστε να αποφύγουν συμπληρωματική εκτομή του ορθού αν υποτροπιάσουν μετά από απλή κολεκτομή και ειλεο-ορθική αναστόμωση.
Άλλοι παράγοντες που πρέπει να λαμβάνονται υπόψη είναι η γονιμότητα και η ανάπτυξη δεσμοειδών όγκων. Η πρωκτοκολεκτομή έχει συσχετισθεί με μεγαλύτερα ποσοστά υπογονιμότητας σε γυναίκες με πολυποδίαση και γι΄αυτό πρέπει να αποφεύγεται σε νεαρές γυναίκες αναπαραγωγικής ηλικίας που επιθυμούν να τεκνοποιήσουν. Σε ασθενείς με ιστορικό δεσμοειδών όγκων ή μεταλλαγή στο κωδικόνιο 1444 του APC πρέπει να επιλέγεται εξαρχής η πρωκτοκολεκτομή καθώς έχουν μεγάλη πιθανότητα ανάπτυξης τέτοιων όγκων και η μετατροπή από απλή κολεκτομή και ειλεο-ορθική αναστόμωση σε πρωκτοκολεκτομή μπορεί να είναι δύσκολη λόγω βράχυνσης του μεσεντερίου (9). Παρόλα αυτά, άλλοι ερευνητές πιστεύουν ότι η μεταλλαγή στο 1444 μπορεί να σχετίζεται και με ήπιο φαινότυπο και η πρωκτοκολεκτομή να είναι υπερβολική. Οι ασθενείς με πρωκτοκολεκτομή μπορεί να αναπτύξουν αδενώματα ή ακόμα και καρκίνο στη ειλεϊκή λήκυθο, και γι΄αυτό απαιτείται παρακολούθηση ανά 6-12 μήνες (10).
Αν και τα μη στεροειδή αντιφλεγμονώδη φάρμακα δεν αντικαθιστούν τη χειρουργική αντιμετώπιση του συνδρόμου της οικογενούς αδενωματώδους πολυποδίασης, εντούτοις μπορεί να παίζουν σημαντικό ρόλο στην αναβολή της χειρουργικής επέμβασης ασθενών με ήπια πολυποδίαση του παχέος εντέρου ή ασθενών με πολυποδίαση του ορθού μετά από προηγούμενη κολεκτομή. Μπορούν επίσης να χρησιμοποιηθούν σε ασθενείς που δεν επιθυμούν χειρουργική επέμβαση ή σε εκείνους με εκτεταμένους δεσμοειδείς όγκους. Το πρώτο φάμακο που αποδείχτηκε αποτελεσματικό στους ασθενείς αυτούς ήταν η σουλινδάκη (11). Η μακροχρόνια θεραπεία με σουλινδάκη ελαττώνει τον αριθμό των αδενωμάτων του παχέος εντέρου περισσότερο από 50% καθώς και του υπολειπόμενου τμήματος ορθού στους ασθενείς που έχουν υποβληθεί σε κολεκτομή και ειλεο-ορθική αναστόμωση.
Το 1990 αναπτύχθηκαν οι εκλεκτικοί COX-2 (cyclo-oxygenase-2) αναστολείς που εμφάνιζαν λιγότερες ανεπιθύμητες ενέργειες εκ του γαστρεντερικού από τα κλασσικά μη στερεοειδή αντιφλεγμονώδη φάρμακα. Ένας από αυτούς τους αναστολείς, η σελεκοξίμπη, βρέθηκε να ελαττώνει κατά 28% τον αριθμό των αδενωμάτων του παχέος εντέρου και του ορθού (12). Σε αντίθεση με τη σουλινδάκη, η σελεκοξίμπη μειώνει ακόμα και τον αριθμό των αδενωμάτων του δωδεκαδακτύλου.
Αδενωματώδης πολυποδίαση σχετιζόμενη με το γονίδιο MUTYH
Το 2002 αναφέρθηκε η σημασία της ανεπαρκούς επιδιόρθωσης βάσεων του DNA στον κληρονομικό καρκίνο του παχέος εντέρου και ορθού (5). Oμόζυγες μεταλλαγές του γονιδίου MUTYH έχουν αναδειχθεί στο 26-29% των ασθενών με 10-100 πολύποδες και στο 7-29% των ασθενών με 100-1000 πολύποδες (13). Ασθενείς με περισσότερους από 10 πολύποδες πρέπει να παραπέμπονται για γενετική καθοδήγηση και να πραγματοποιείται έλεγχος μεταλλαγών του γονιδίου MUTYH. Οι μεταλλαγές του συγκεκριμένου γονιδίου συνήθως σχετίζονται με ήπιο φαινότυπο πολυποδίασης. Μέχρι σήμερα, κακοήθεια του εντέρου και εξωτεντερικές αλλοιώσεις σχετιζόμενες με οικογενή αδενωματώδη πολυποδίαση όπως είναι ο δωδεκαδακτυλικός καρκίνος, οστεώματα και συγγενής υπετροφία του χρωστικού επιθηλίου του αμφιβληστροειδούς έχουν μόνο σποραδικά αναφερθεί σε ασθενείς με πολυποδίαση σχετιζόμενη με το MUTYH. Το προτεινόμενο πρωτόκολλο παρακολούθησης ασθενών με σχετιζόμενη με μεταλλαγές του MUTYH πολυποδίαση είναι παρόμοιο με αυτό των ασθενών με την ήπια μορφή του συνδρόμου της οικογενούς πολυποδίασης.
Όσον αφορά την καταλληλότερη χειρουργική αντιμετώπιση της πολυποδίασης σε αυτούς τους ασθενείς πρέπει αν σημειωθεί οτι ο ήπιος φαινότυπος με μικρό αριθμό αδενωμάτων καθιστά δυνατή σε αρκετούς ασθενείς την ενδοσκοπική αφαίρεση των πολυπόδων. Αν απαιτείται μείζονα χειρουργική επέμβαση, στις περισσότερες των περιπτώσεων αυτή είναι η ολική κολεκτομή και ειλεο-ορθική αναστόμωση για να περιοριστεί ο κίνδυνος ανάπτυξης καρκίνου. Στην περίπτωση που υπάρχει σοβαρή πολυποδίαση στο ορθό προτείνεται ολική πρωκτοκολεκτομή και ειλεο-πρωκτική αναστόμωση.
Σύνδρομο Lynch
Οι ασθενείς που φέρουν μεταλλαγή στα γονίδια επιδιόρθωσης του DNA εμφανίζουν υψηλό κίνδυνο ανάπτυξης καρκίνου του παχέος εντέρου, του ενδομητρίου και μιας σειράς άλλων νεοπλασμάτων που συνοψίζονται στον Πίνακα 3 (14, 15). Οι καρκίνοι αυτοί εντοπίζονται σε νεαρή ηλικία και μπορεί να είναι σύγχρονοι.

Πίνακας 3:
Δια βίου κίνδυνος ανάπτυξης διαφόρων τύπων καρκίνου σε ασθενείς με μεταλλαγή στα γονίδια επιδιόρθωσης του DNA
Η βλάβη στα γονίδια επιδιόρθωσης οδηγεί κατά την αντιγραφή σε λάθη σε επαναλαμβανόμενες αλληλουχίες του DNA που καλούνται μικροδορυφορικό DNA. Το φαινόμενο αυτό ονομάζεται μικροδορυφορική αστάθεια και παρατηρείται στο 90% των καρκίνων του παχέος εντέρου που σχετίζονται με το σύνδρομο Lynch και μόλις στο 15% των σποραδικών καρκίνων. Με ανοσοϊστοχημική ανάλυση των πρωτεϊνών που παράγονται από τα γονίδια επιδιόρθωσης μπορεί να αναδειχθεί απώλεια της έκφρασης και να εντοπισθεί το υπεύθυνο γονίδιο.
Το 1989 διατυπώθηκαν στο Άμστερνταμ ορισμένα κριτήρια ένταξης των συγκεκτιμένων οικογενειών στο σύνδρομο Lynch (16). Τα κριτήρια αυτά αναθεωρήθηκαν τo 1999 ώστε να περιλαμβάνουν και τις εξωεντερικές εκδηλώσεις της νόσου (17). Το 1997 προτάθηκαν τα κριτήρια της Bethesda με στόχο να καθορίζονται τα άτομα που πρέπει να υποβάλλονται σε έλεγχο μικροδορυφορικής αστάθειας (18, 19). Τα κριτήρια αυτά αναθεωρήθηκαν το 2004 (20). Με βάση τα κριτήρια αυτά έχει αναγνωρισθεί μια μεγάλη ομάδα οικογενειών που εντάσσονται στο σύνδρομο Lynch. Πρέπει να σημειωθεί ότι η ονομασία κληρονομικός μη πολυποδιακός καρκίνος του παχέος εντέρου (Hereditary Non Polyposis Colorectal Cancer – HNPCC) που προτάθηκε στο Άμστερνταμ το 1989 σταδιακά εγκαταλείφθηκε, καθώς δεν περιελάμβανε τους όγκους σε άλλα όργανα που συχνά παρατηρούνται. Έτσι καθιερώθηκε η ονομασία σύνδομο Lynch.
Σήμερα, για την επιλογή των ασθενών με καρκίνο παχέος εντέρου που θα ελεγχθούν για ύπαρξη μοριακών αλλαγών ή εκείνων με μικροδορυφορική αστάθεια που θα ελεγχθούν για μεταλλαγές στα γονίδια επιδιόρθωσης χρησιμοποιούνται τα κριτήρια του Άμστερνταμ ΙΙ ή τα αναθεωρημένα κριτήρια της Bethesda (Πίνακες 4, 5).

Πίνακας 4:
Κριτήρια του Άμστερνταμ ΙΙ

Πίνακας 5:
Αναθεωρημένα κριτήρια της Bethesda
Ο αριθμός των χρησιμοποιούμενων δεικτών μικροδορυφορικής αστάθειας ποικίλλει από 1 έως άνω των 10. Για την ανοσοϊστοχημεία χρησιμοποιούνται κυρίως δύο αντισώματα (MLH1 και MSH2), αλλά υπάρχουν και μελέτες που χρησιμοποιούν 3 ή 4 αντισώματα (MLH1, MSH2 MSH6 και PMS2). Στις μελέτες όπου εφαρμόστηκαν ταυτόχρονα και προοπτικά τόσο η μικροδορυφορική αστάθεια όσο και η ανοσοϊστοχημεία, η ευαισθησία της πρώτης αποδείχθηκε ελαφρώς μεγαλύτερη από τη δεύτερη. Το πλεονέκτημα της ανοσοϊστοχημείας είναι ότι μπορεί να υποδεικνύει την ακριβή γονιδιακή βλάβη από το είδος της παρατηρούμενης χρώσης. Για αυτό το λόγο οι περισσότεροι ερευνητές προτείνουν τη μέθοδο αυτή ως τη βασική για τον πρωταρχικό έλεγχο οικογενειών με πιθανή μεταλλαγή στα γονίδια επιδιόρθωσης του DNA (π.χ. οικογένειες που καλύπτουν τα κριτήρια του Άμστερνταμ). Λόγω της μεγαλύτερης ευαισθησίας της μικροδορυφορικής αστάθειας, αυτή προτείνεται σε περιπτώσεις με υψηλή πιθανότητα συνδρόμου Lynch αλλά φυσιολογική παρατηρούμνεη έκφραση των πρωτεϊνών επιδιόρθωσης. Στις υπόλοιπες περιπτώσεις έγκειται στην εμπειρία του κάθε κέντρου για το ποιά μέθοδος ανίχνευσης βλαβών στα γονίδια επιδιόρθωσης θα εφαρμοστεί (Σχήμα 1).
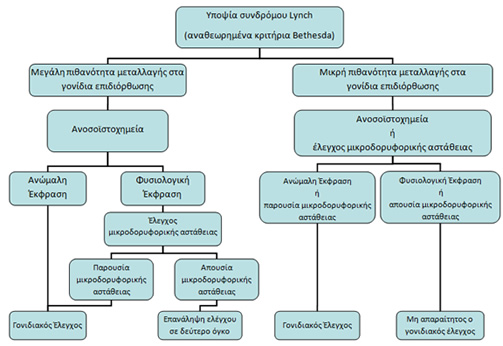
Σχήμα 1.
Στρατηγική αναγνώρισης ασθενών με καρκίνο του παχέος εντέρου και μεταλλαγή στα γονίδια επιδιόρθωσης του DNA
Η παρακολούθηση των ασθενών του συνδρόμου πρέπει να ξεκινά μεταξύ 20 και 25 ετών και να συνεχίζεται δια βίου εάν η κατάσταση της υγείας του ατόμου το επιτρέπει. Όσον αφορά τα μεσοδιαστήματα παρακολούθησης του εντέρου στο σύνδρομο Lynch, έχει αποδειχθεί ότι η εξέταση ανά τριετία είναι σε μεγάλο βαθμό αποτελεσματική. Δεδομένου όμως των προχωρημένων σταδίων του καρκίνου που εντοπίζεται στις συγκεκριμένες οικογένειες προτείνεται η παρακολούθηση να γίνεται ανά 1-2 έτη. Η αξία της παρακολούθησης για τον καρκίνο του ενδομητρίου δεν μπορεί ακόμα με ακρίβεια να προσδιοριστεί. Η γυναικολογική εξέταση, το διακολπικό υπερηχογράφημα και η βιοψία από την ηλικία των 30 ως 35 ετών μπορεί να οδηγήσουν στη διαγνωση προκαρκινικών αλλοιώσεων και πρώιμου σταδίου καρκίνων. Η προφυλακτική υστερεκτομή και σαλπιγγο-ωοθηκεκτομή αποτελούν αξιόλογες επιλογές για τις γυναίκες με σύνδρομο Lynch καθώς ελαττώνουν δραστικά την ανάπτυξη καρκίνου στα συγκεκριμένα όργανα.
Στον Πίνακα 6 συνοψίζονται οι κατευθυντήριες οδηγίες παρακολούθησης των οικογενειών με σύνδρομο Lynch. Οι οδηγίες αυτές έχουν ένδειξη σε οικογένειες με γνωστή μεταλλαγή στα γονίδια επιδιόρθωσης καθώς και σε οικογένειες με συσσώρευση περιστατικών καρκίνου παχέος εντέρου ή άλλων σχετιζόμενων καρκίνων και υποψία βλάβης του συστήματος επιδιόρθωσης (παρουσία μικροδορυφορικής αστάθειας ή απώλεια της έκφρασης των γονιδίων επιδιόρθωσης).
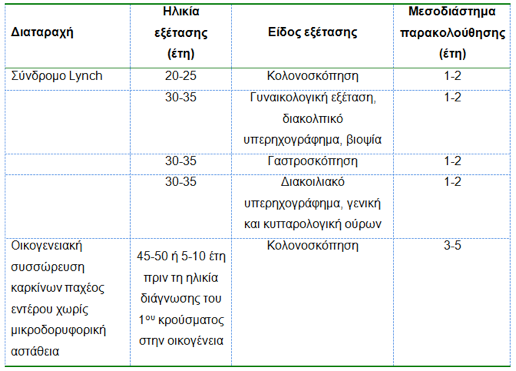
Πίνακας 6:
Πρωτόκολλο παρακολούθησης ασθενών με σύνδρομο Lynch ή οικογενειών με συσσώρευση κρουσμάτων καρκίνου παχέος εντέρου
Πολλαπλές μελέτες έχουν δείξει ότι ασθενείς με σύνδρομο Lynch έχουν αυξημένο κίνδυνο ανάπτυξης σύγχρονων και μετάχρονων καρκίνων του παχέος εντέρου. Γι’ αυτό το λόγο είναι σημαντικός ο έλεγχος ολόκληρου του παχέος εντέρου πριν την εκτομή ενός όγκου καθώς υπάρχει κίνδυνος ύπαρξης σύγχρονου όγκου. Λόγω του αυξημένου κινδύνου ανάπτυξης δεύτερου όγκου πρέπει να εξετάζεται το ενδεχόμενο της υφολικής κολεκτομής και ειλεο-ορθικής αναστόμωσης σε νέους ασθενείς.
Οι μελέτες δείχνουν ότι δεν υπάρχει όφελος σε αυτούς τους ασθενείς από τη θεραπεία με 5-φθοριο-ουρακίλη. Μία μικρή μελέτη σε ασθενείς με καρκίνο παχέος εντέρου σταδίου IV αναφέρει πλήρη ή μερική απάντηση στη θεραπεία με ιρινοτεκάνη.
Ενδεικτική βιβλιογραφία
1. Vasen HF, Moslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007;44:353–62.
2. Bulow S. Results of national registration of familial adenomatous polyposis. Gut 2003;52:742–6.
3. Cruz-Correa M, Giardiello FM. Familial adenomatous polyposis. Gastrointest Endosc 2003;58:885–94.
4. Nielsen M, Hes FJ, Nagengast FM, et al. Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis. Clin Genet 2007;71:427–33.
5. Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:CRT:A mutations in colorectal tumors. Nat Genet 2002;30:227–32.
6. Aretz S, Uhlhaas S, Caspari R, et al. Frequency and parental origin of de novo APC mutations in familial adenomatous polyposis. Eur J Hum Genet 2004;12:52–8.
7. Aziz O, Athanasiou T, Fazio VW, et al. Meta-analysis of observational studies of ileorectal versus ileal pouch–anal anastomosis for familial adenomatous polyposis. Br J Surg 2006;93:407–17.
8. Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol 2007;61:153–61.
9. Olsen KO, Juul S, Bulow S, et al. Female fecundity before and after operation for familial adenomatous polyposis. Br J Surg 2003;90:227–31.
10. Wu JS, McGannon EA, Church JM. Incidence of neoplastic polyps in the ileal pouch of patients with familial adenomatous polyposis after restorative proctocolectomy. Dis Colon Rectum 1998;41:552–6.
11. Cruz-Correa M, Hylind LM, Romans KE, et al. Long-term treatment with sulindac in familial adenomatous polyposis: a prospective cohort study. Gastroenterology 2002;122:641–5.
12. Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000;342:1946–52.
13. Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med 2003;348:791–9.
14. Lynch HT, Chapelle de la A. Hereditary colorectal cancer. N Engl J Med 2003;348:919–32.
15. Al Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:CRT:A mutations in colorectal tumors. Nat Genet 2002;30:227–32.
16. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991;34:424–5.
17. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999;116:1453–6.
18. Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin L, Srivastava S. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997;89:1758–62.
19. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998;58:5248–57.
20. Umar A, Boland CR, Terdiman JP, Syngal S, de la CA, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261–8.
